# Archivos descargados de cf.10xgenomics.com:
# 1. HumanColonCancer_Flex_Multiplex_count_filtered_feature_bc_matrix.tar.gz
# 2. HumanColonCancer_Flex_Multiplex_count_analysis.tar.gz
# Después de descomprimir:
# filtered_feature_bc_matrix/ ← matriz de conteo (MTX)
# ├── barcodes.tsv.gz (278,610 barcodes con sufijo -1 a -32)
# ├── features.tsv.gz
# └── matrix.mtx.gz
# analysis/
# └── umap/gene_expression_2_components/projection.csvClase 4: Deconvolución e Integración con scRNA-seq
2026-05-20
UMAP de Cell Ranger: ¿Pacientes separados?
Code
# Cell Ranger calcula un UMAP durante el pipeline aggr
umap_cr <- read.csv("flex_scrna/analysis/umap/gene_expression_2_components/projection.csv")
rownames(umap_cr) <- umap_cr$Barcode
umap_cr <- umap_cr[colnames(flex), ]
flex[["umap"]] <- CreateDimReducObject(
embeddings = as.matrix(umap_cr[, c("UMAP.1", "UMAP.2")]),
key = "UMAP_", assay = "RNA"
)
DimPlot(flex, group.by = "patient", reduction = "umap", pt.size = 1)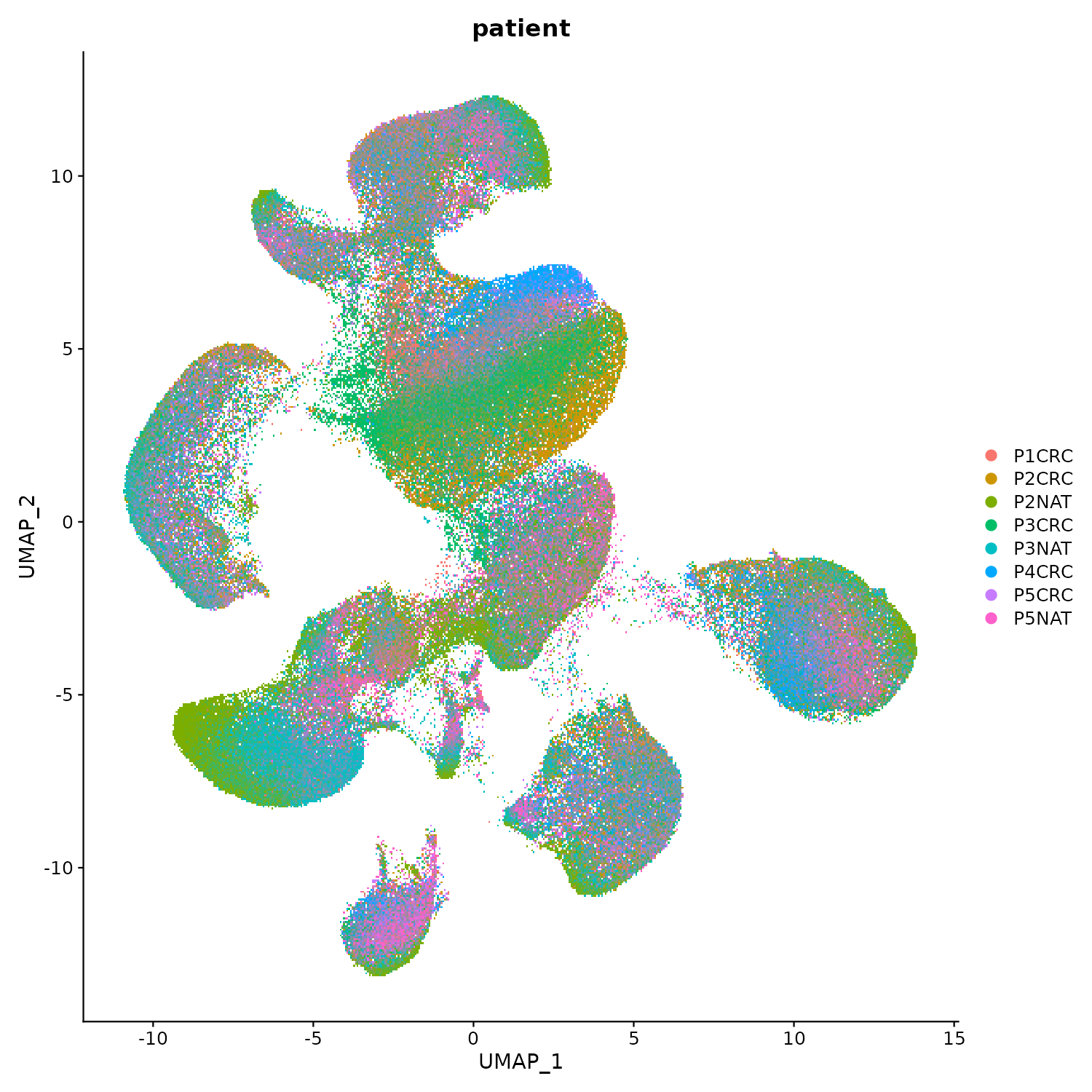
Procesamiento estándar con Seurat
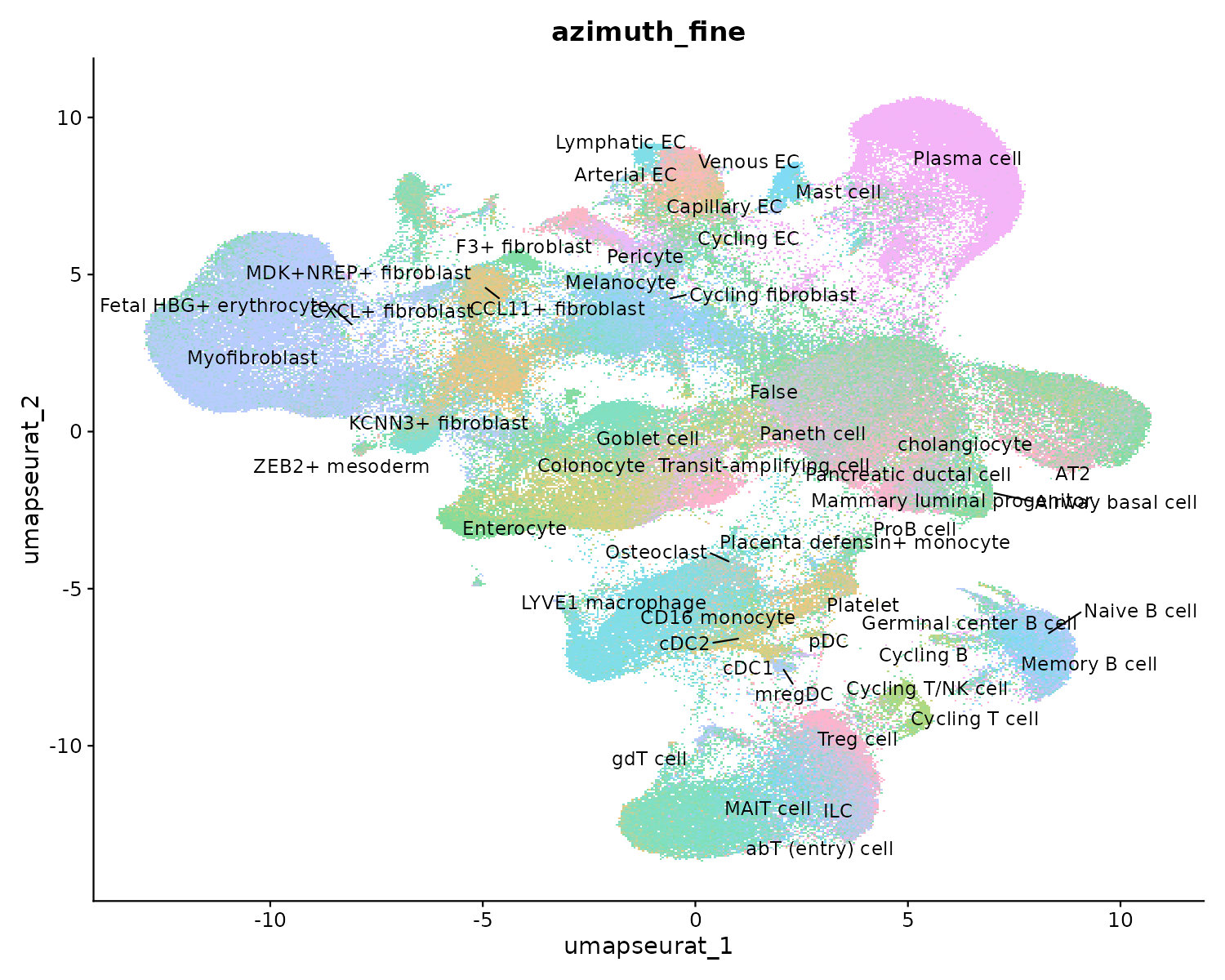
Resultado de Azimuth: anotación fina
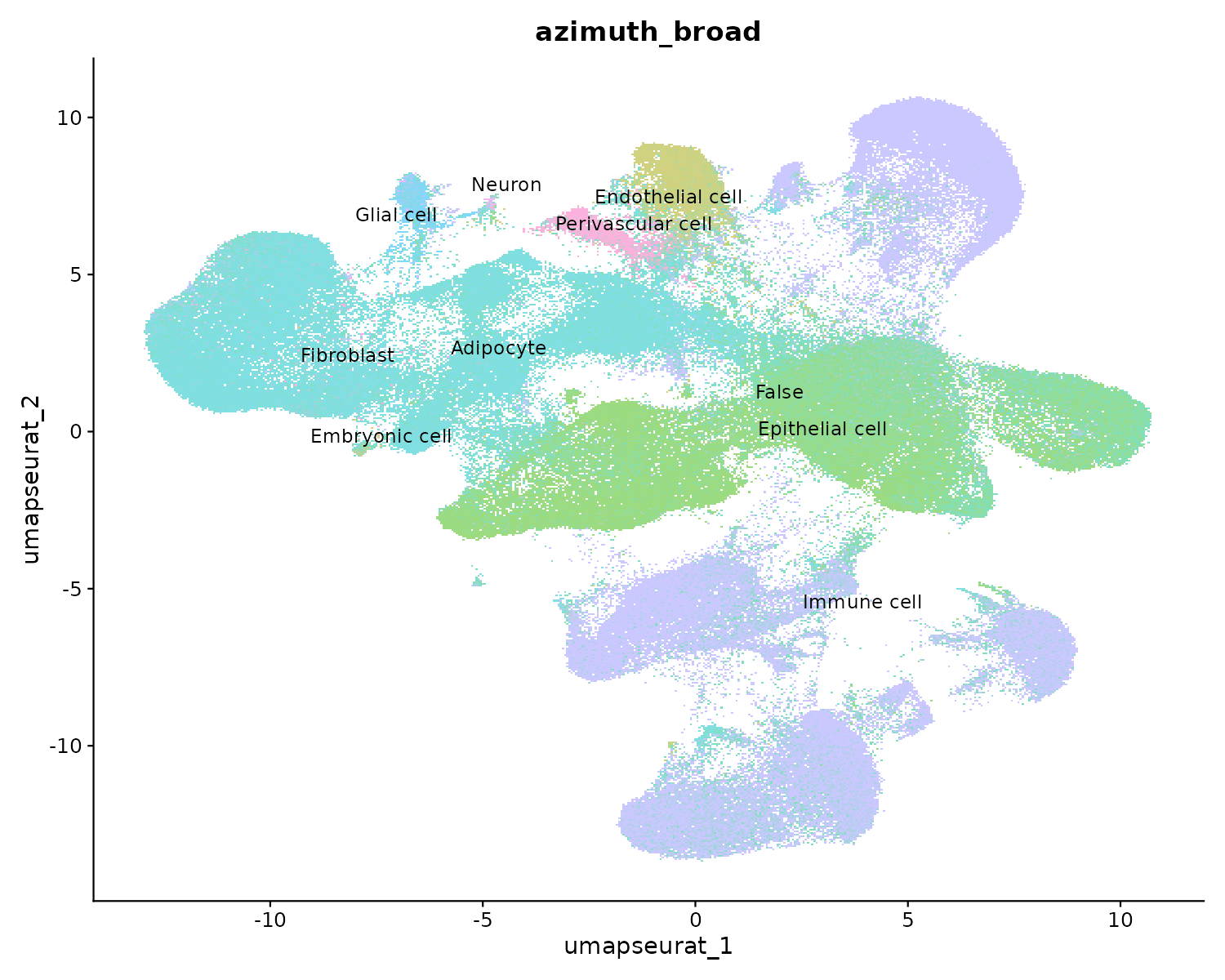
Resultado de Azimuth: anotación amplia
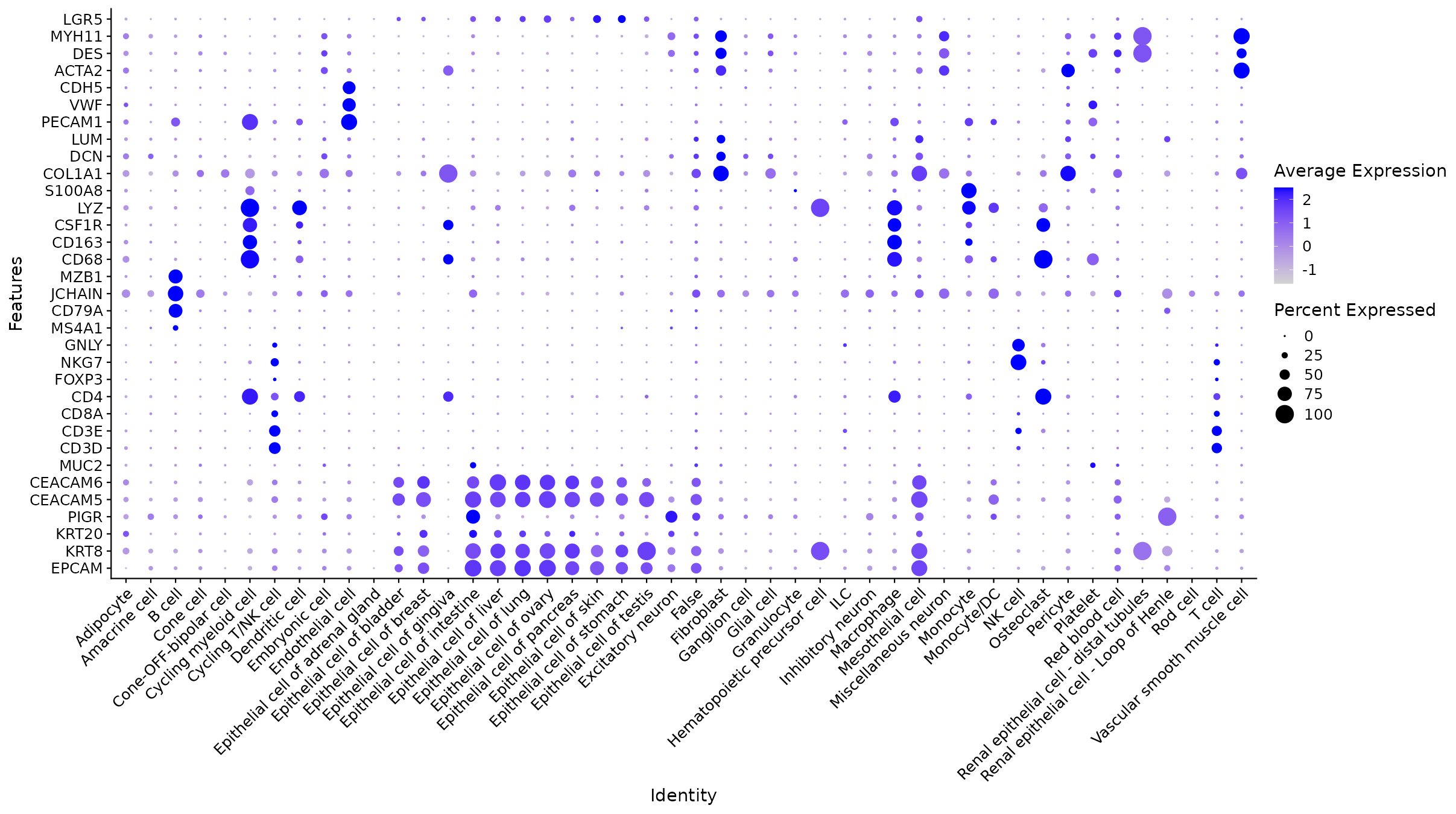
DotPlot: marcadores vs. tipos celulares
FeaturePlot: expresión de marcadores en UMAP

Confianza de la anotación
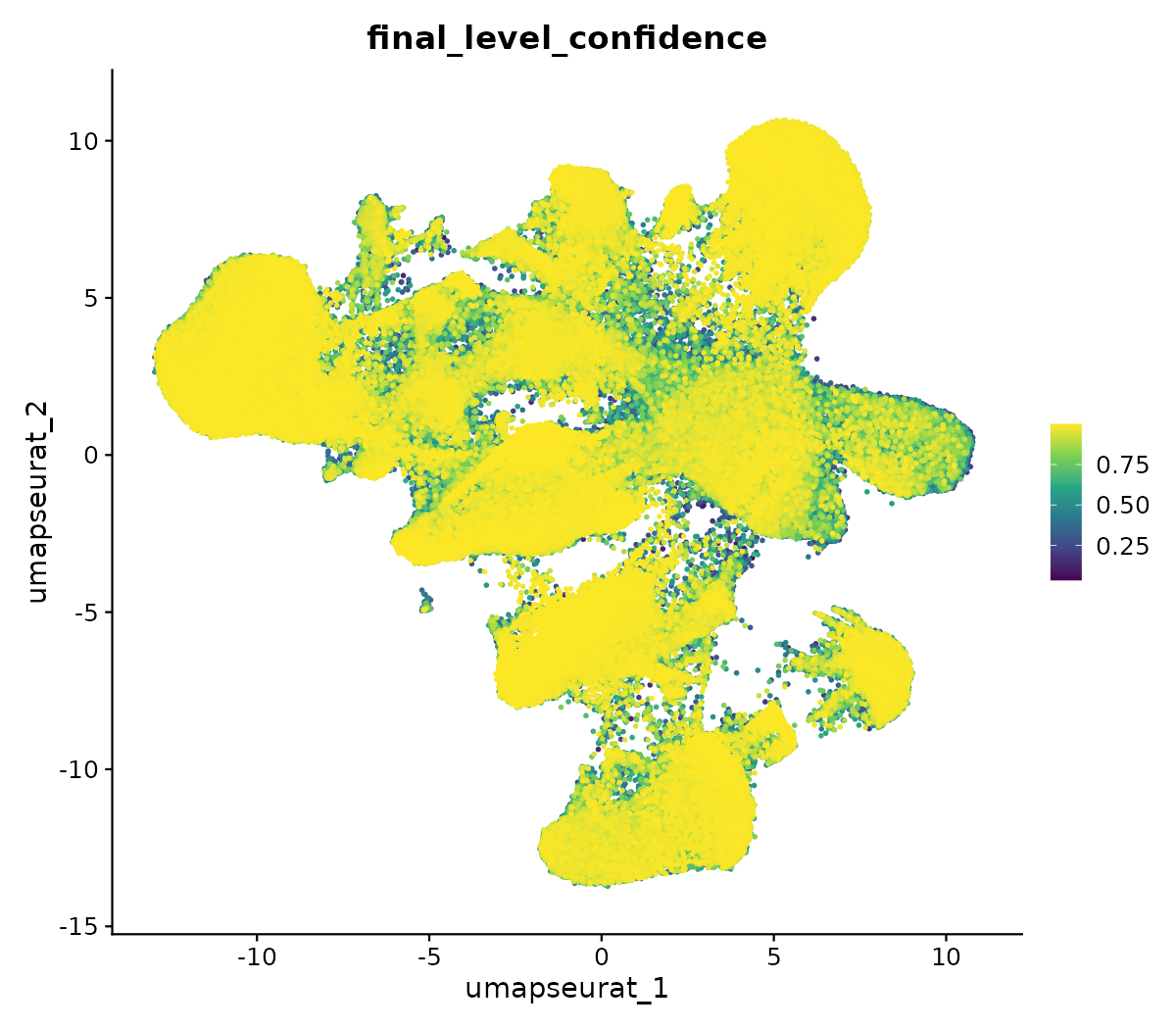
El final_level_confidence indica qué tan segura es la asignación:
- > 0.8: alta confianza
- 0.5–0.8: aceptable
- < 0.5: revisar manualmente
Las células con baja confianza suelen estar en:
- Transiciones entre tipos
- Tipos no representados en la referencia
- Doublets / baja calidad
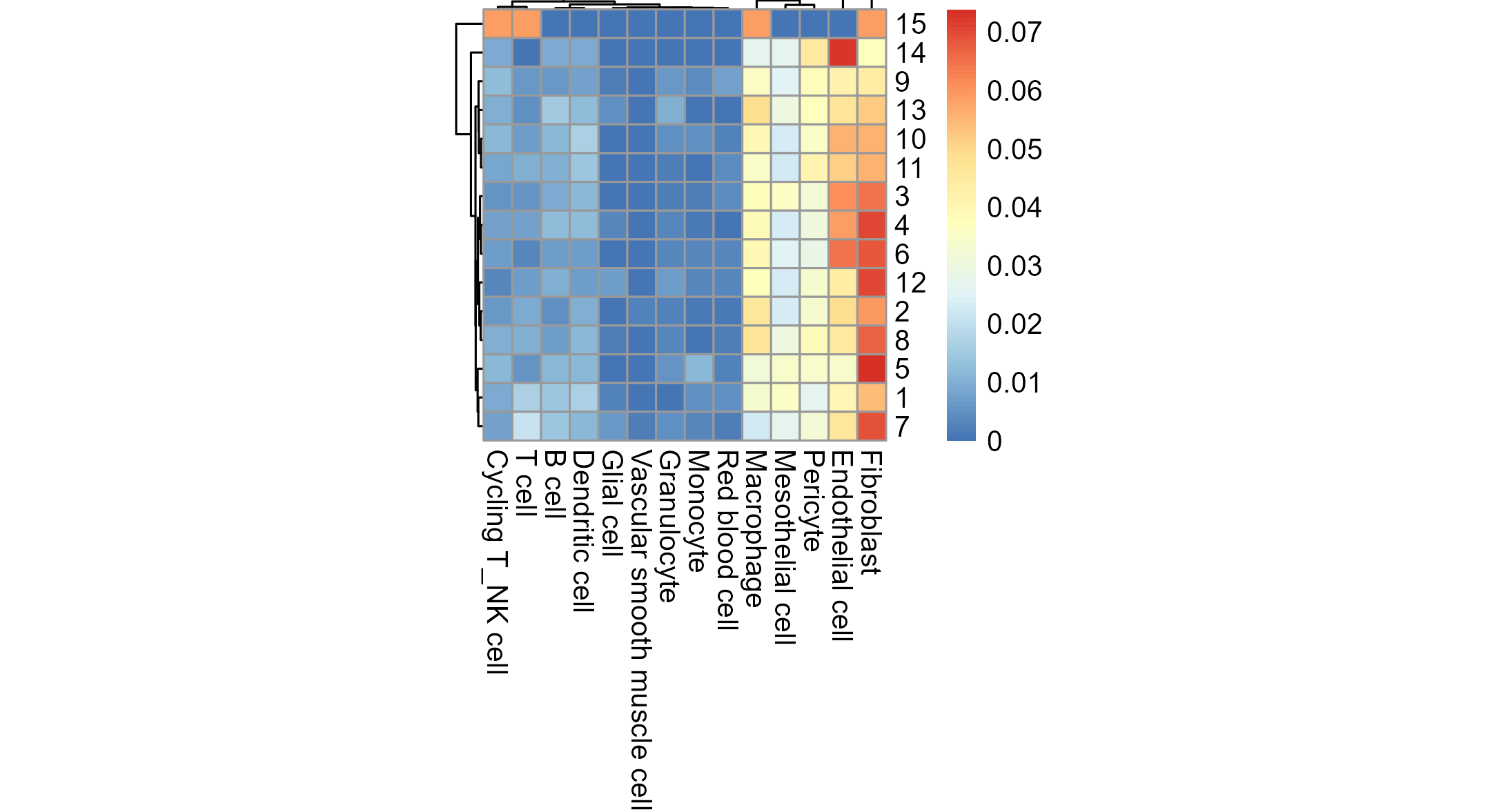
Cluster vs. tipo celular predicho
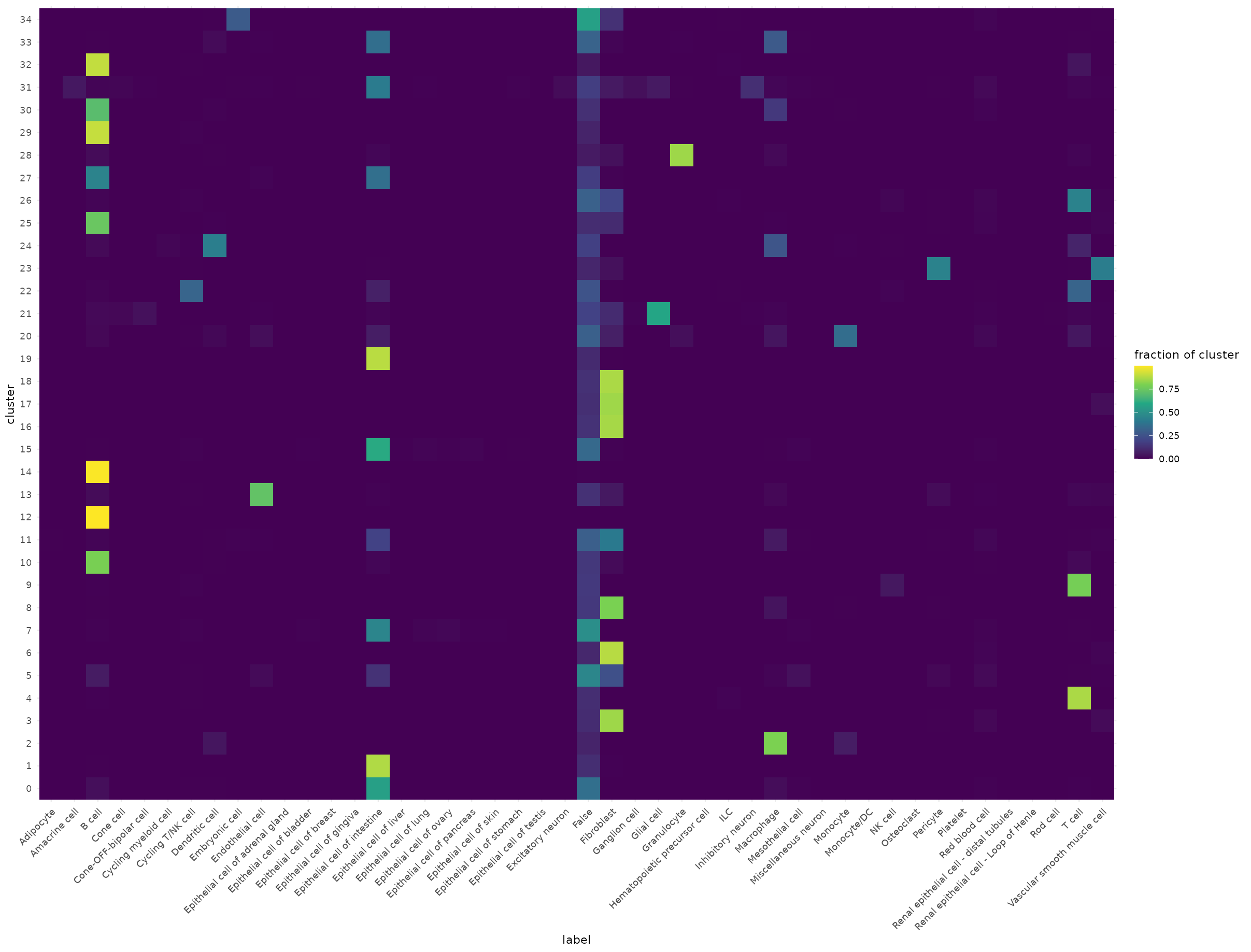
Cada fila es un cluster de Seurat, cada columna un tipo celular predicho por Azimuth. Si un cluster tiene una sola columna dominante, la anotación es consistente.
¿Qué es la deconvolución espacial?
Cada bin de Visium HD (8 µm) puede contener 1–2 células. El perfil de expresión del bin es una mezcla de los tipos celulares presentes.
Deconvolución = estimar la composición de tipos celulares por bin, usando el atlas scRNA-seq como referencia.
Herramienta: spacexr / RCTD
- Robust Cell Type Decomposition
- Modelo probabilístico que estima proporciones
- Modo
doublet: asume máximo 2 tipos por bin (ideal para 8 µm)

Subsetting opcional para que no tarde tanto
Code
count.plot <- SpatialFeaturePlot(object, features = "nCount_Spatial") +
theme(legend.position = "right")
# Extract everything we need
plot_data <- ggplot_build(count.plot)$data[[1]]
x_cuts <- seq(min(plot_data$x), max(plot_data$x), length.out = 9)
y_cuts <- seq(min(plot_data$y), max(plot_data$y), length.out = 9)
ggplot(plot_data, aes(x = x, y = y, color = fill)) +
geom_point(size = 0.1, shape = 15) +
scale_color_identity() +
# Grid
annotate("segment",
x = x_cuts, xend = x_cuts,
y = min(plot_data$y), yend = max(plot_data$y),
color = "white", linewidth = 0.3, linetype = "dashed") +
annotate("segment",
x = min(plot_data$x), xend = max(plot_data$x),
y = y_cuts, yend = y_cuts,
color = "white", linewidth = 0.3, linetype = "dashed") +
# Column labels
annotate("text",
x = head(x_cuts, -1) + diff(x_cuts) / 2,
y = rep(max(plot_data$y) + 5, 8),
label = 1:8, color = "yellow", size = 3, fontface = "bold") +
# Row labels
annotate("text",
x = rep(min(plot_data$x) - 5, 8),
y = head(y_cuts, -1) + diff(y_cuts) / 2,
label = LETTERS[1:8], color = "yellow", size = 3, fontface = "bold") +
coord_fixed(clip = "off") +
theme_void() +
theme(legend.position = "none",
plot.background = element_rect(fill = "black", color = NA))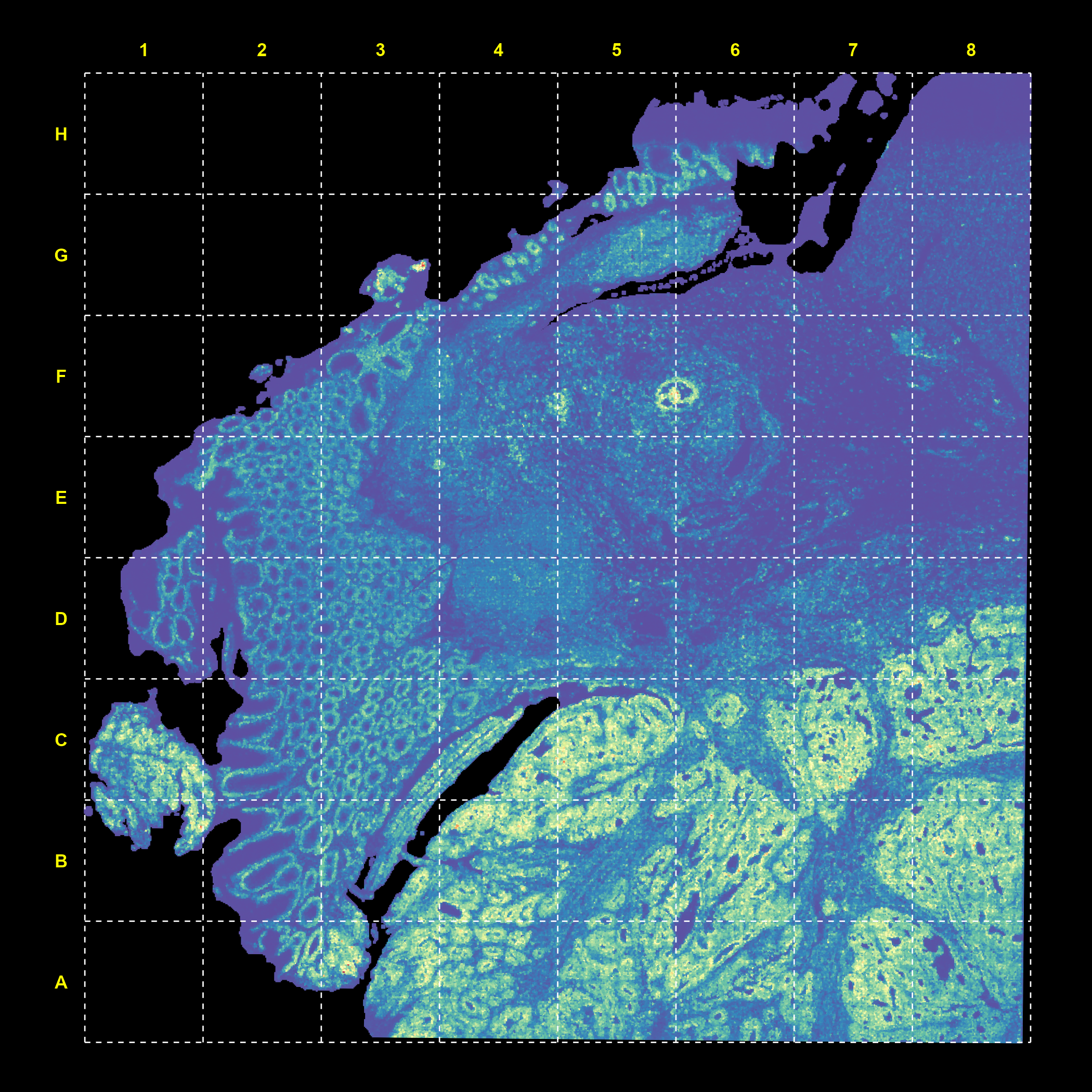

Visualizando la deconvolución
Code
# Extraer los resultados
rctd <- readRDS("resultsRCTD_P1CRC.rds")
results <- rctd@results
# Para modo doublet: tipo celular 1 y 2 por bin + proporciones
cell_type_1 <- results$results_df$first_type
#weirdly stored as scond_type (sic)
cell_type_2 <- results$results_df$scond_type
visium$rctd_class <- results$results_df$spot_class
visium$rctd_weight1 <- results$weights_doublet[, "first_type"]
# Agregar al objeto Seurat
visium$rctd_type1 <- cell_type_1
visium$rctd_type2 <- cell_type_2
# Visualizar el tipo celular dominante
coords <- data.frame(
x = visium$spatial_col,
y = visium$spatial_row,
type = visium$rctd_type1,
class = visium$rctd_class
)
coords_good <- coords[coords$class != "reject", ]
p <- ggplot(coords_good, aes(x = x, y = -y, color = type)) +
geom_point(size = 0.1, shape = 15) +
coord_fixed() +
theme_minimal() +
theme(axis.title = element_blank(), axis.text = element_blank()) +
guides(color = guide_legend(override.aes = list(size = 3))) +
labs(title = "RCTD deconvolution — dominant cell type")
ggsave("rctd_spatial_celltypes.png", p, width = 12, height = 10, dpi = 150)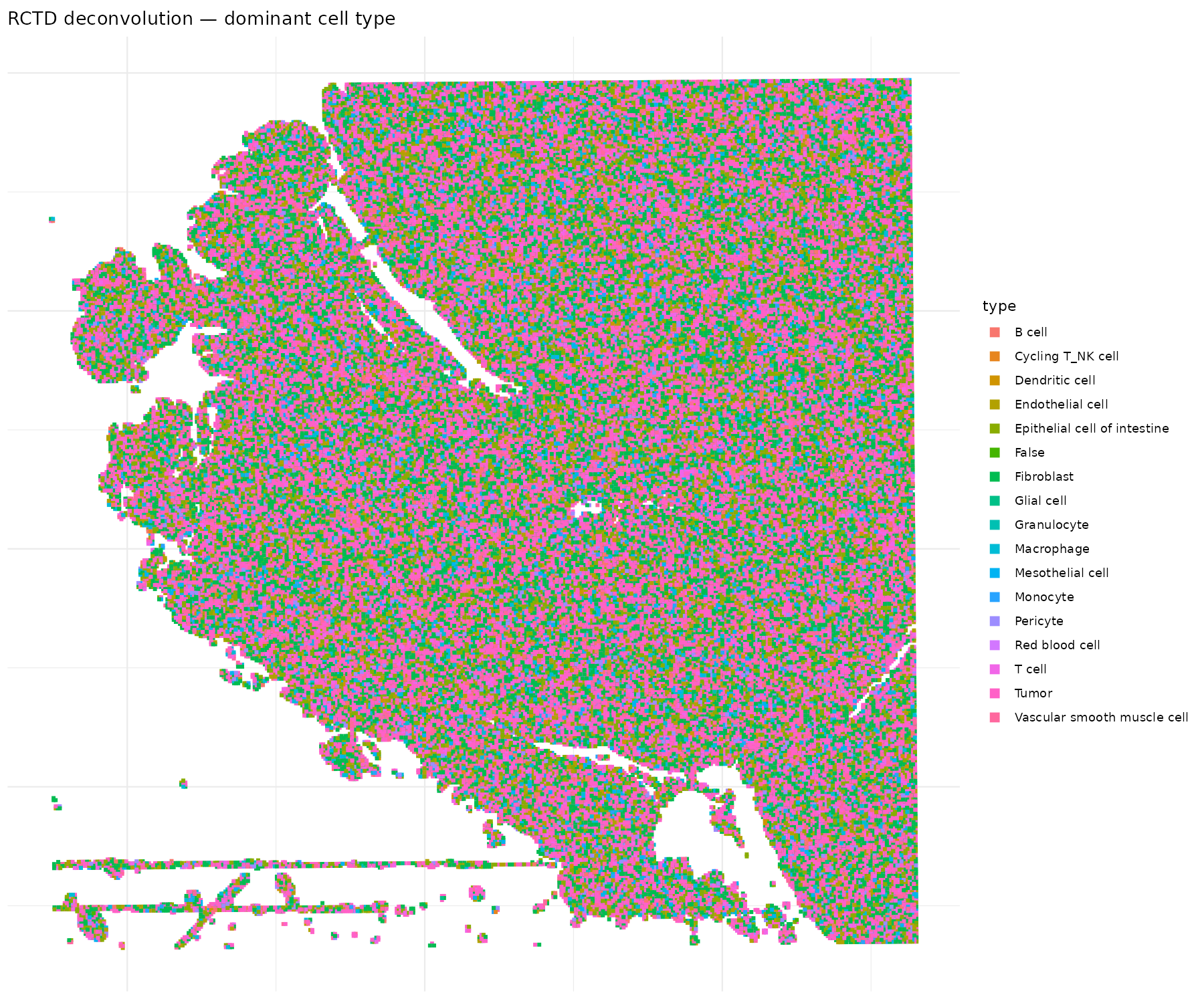
O por paneles
Code
library(patchwork)
weights <- as.data.frame(results$weights)
colnames(weights) <- gsub(" ", "_", colnames(weights)) # spaces break metadata
for (ct in colnames(weights)) {
visium[[ct]] <- weights[[ct]]
}
# Plot helper using your spatial coords
plot_weight <- function(obj, feature) {
df <- data.frame(
x = obj$spatial_col,
y = obj$spatial_row,
val = obj[[feature, drop = TRUE]]
)
ggplot(df, aes(x = x, y = -y, color = val)) +
geom_point(size = 0.3, shape = 15) +
coord_fixed() +
labs(title = feature, color = NULL) +
theme_void() +
theme(plot.title = element_text(size = 9, hjust = 0.5))
}
ct_names <- colnames(weights)
p <- lapply(ct_names, \(ct) plot_weight(visium, ct)) |>
wrap_plots(nrow = 2, guides = "collect") &
scale_color_gradientn(colors = rev(hcl.colors(9, "Rocket"))) &
theme(
legend.key.width = unit(0.5, "lines"),
legend.key.height = unit(1, "lines")
)
ggsave("rctd_all_weights_panels.png", p, width = 20, height = 10, dpi = 150)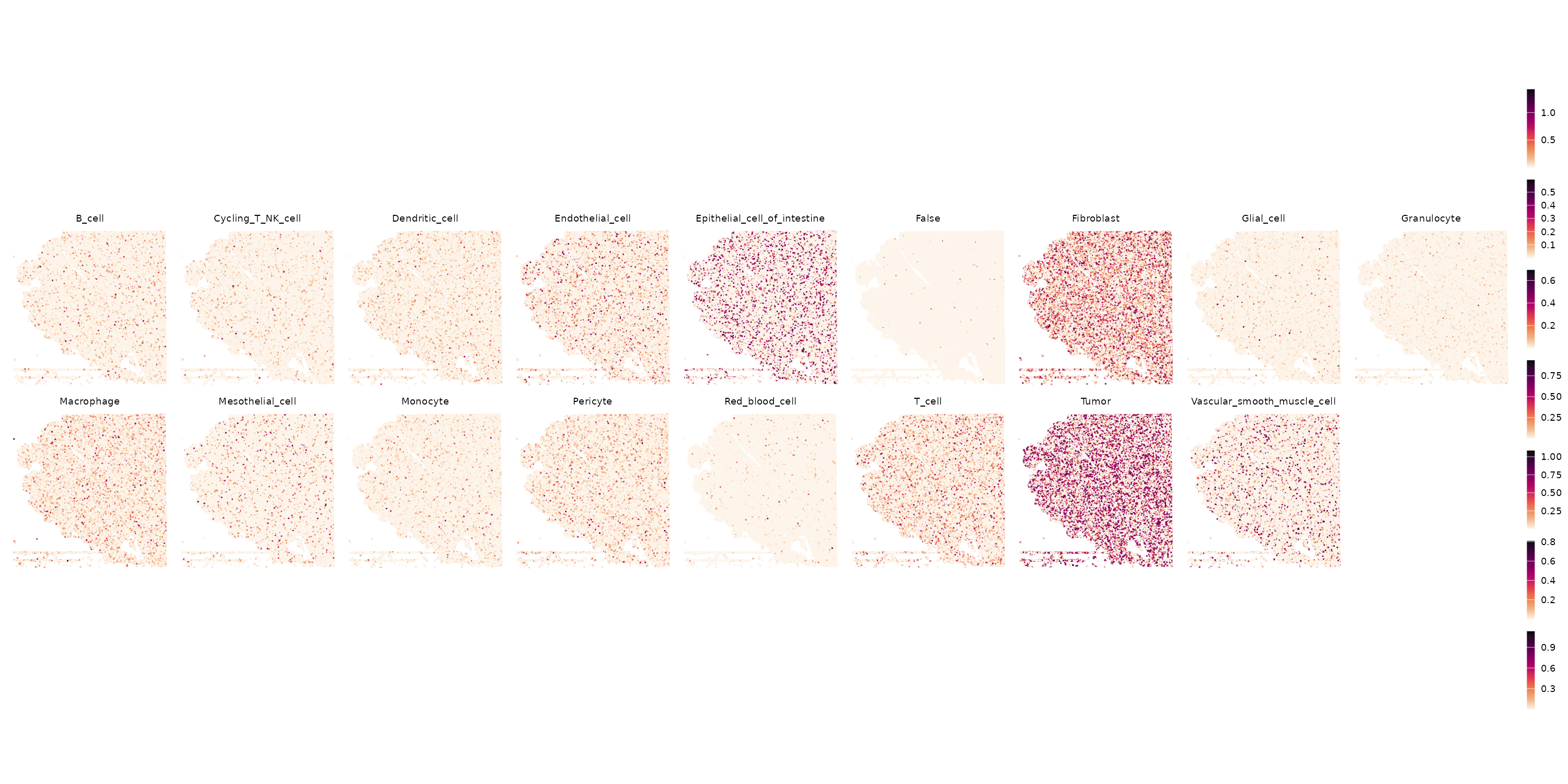
Xenium
Code
df <- data.frame(
x = xenium$x_centroid,
y = xenium$y_centroid,
counts = xenium$nCount_Xenium
)
p <- ggplot(df, aes(x = x, y = -y, color = log1p(counts))) +
geom_point(size = 0.05) +
scale_color_viridis_c() +
coord_fixed() +
theme_void() +
labs(title = "Xenium P1CRC — UMIs per cell")
ggsave("xenium_P1CRC_counts.png", width = 10, height = 8, dpi = 150)