Métodos de Resolución Espacial
Clase 12 — Transcriptómica Espacial


La transcriptómica espacial es la evolución de técnicas histológicas y transcriptómicas
Técnicas histológicas clásicas:
- Hibridación in situ
- Single molecule FISH
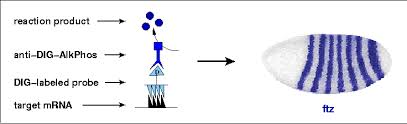
Técnicas transcriptómicas modernas:
- RNA-seq
- scRNA-seq

Motivaciones de la transcriptómica espacial
Form follows function… follows localization
- Identificación de genes con patrones restringidos que indiquen una función en el desarrollo
- Marcadores celulares novedosos y tipos celulares que no son evidentes de la morfología
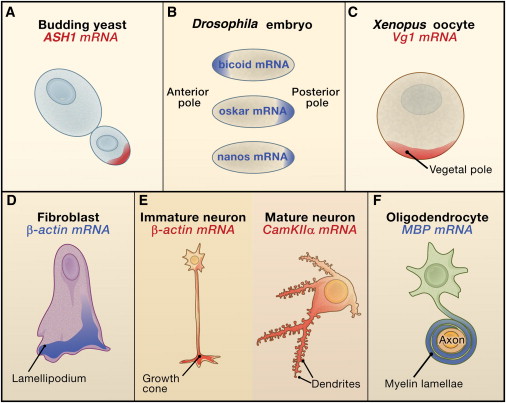
Hasta 70% de los genes expresados en el desarrollo de Drosophila tienen un patrón de expresión
Martin & Ephrussi, 2009
LCM: Laser Capture Microdissection + RNA-seq
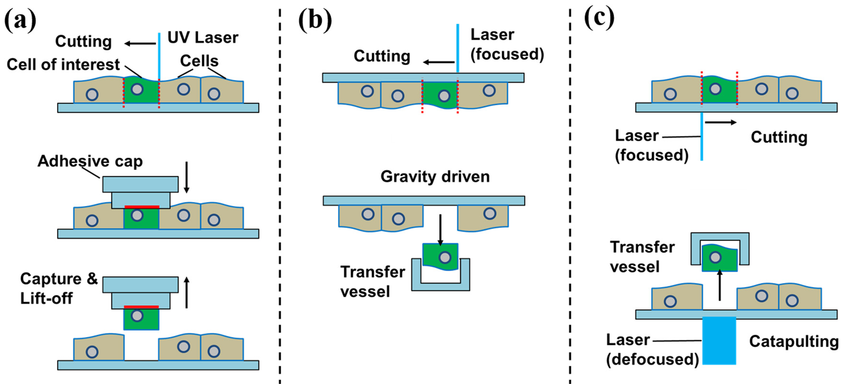
- Se selecciona una región de interés en el tejido mediante láser
- El material se captura sobre un plasma adhesivo
- Se extrae RNA y se realiza RNA-seq estándar
TOMO-seq: RNA-seq en secciones al crióstato
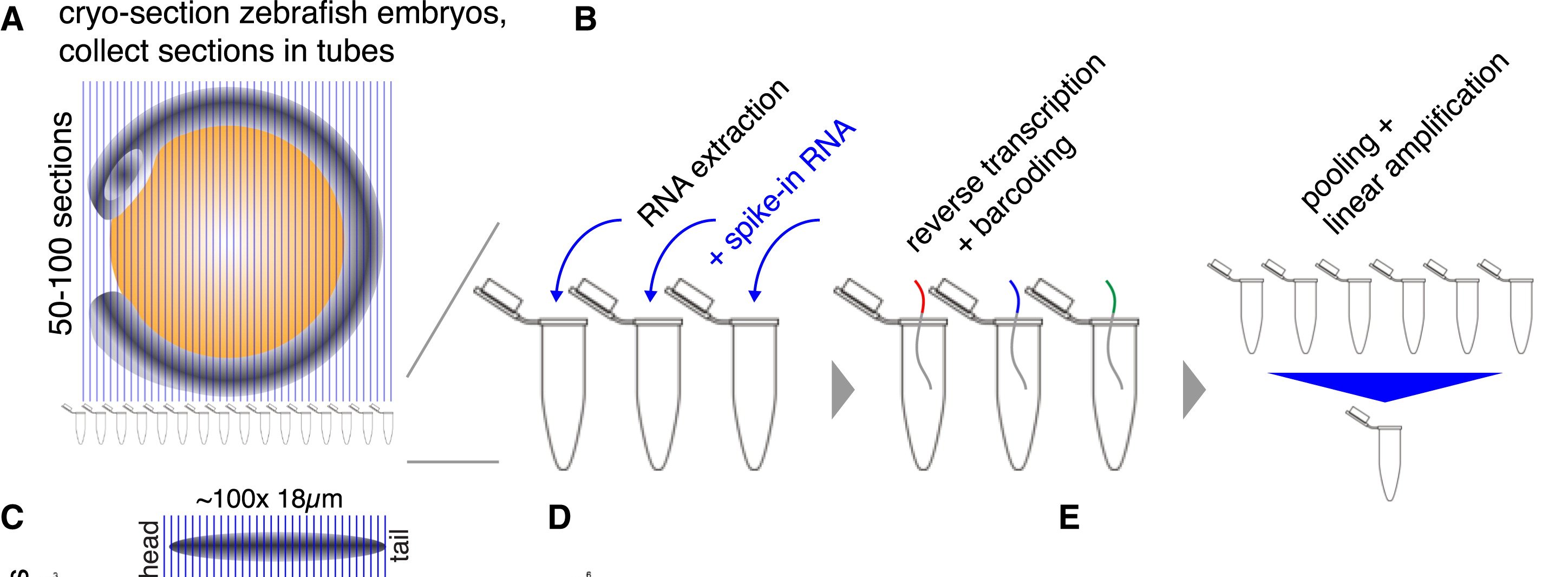
Se secciona el tejido en rebanadas finas y se procesa cada una por separado, con un barcode para luego conjuntarlas en una sóla librería
Transcriptómica espacial 1D, aunque pueden hacerse en las tres orientaciones y reconstruirse.
Niche-seq: fotoactivación y FACS
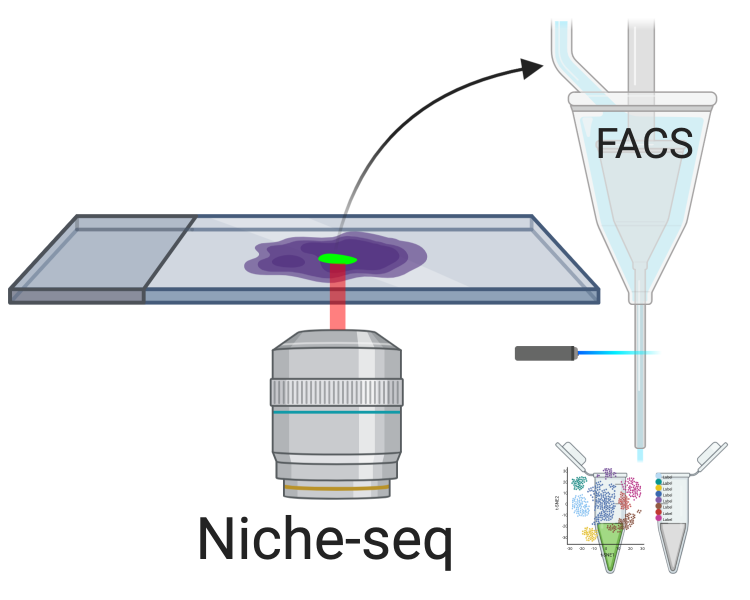
- Se fotoactivan células en una región definida
- Se disocian y clasifican por FACS
- Células fotoactivadas se procesan para RNA-seq
GeomX DSP: Barcodes en Región de Interés
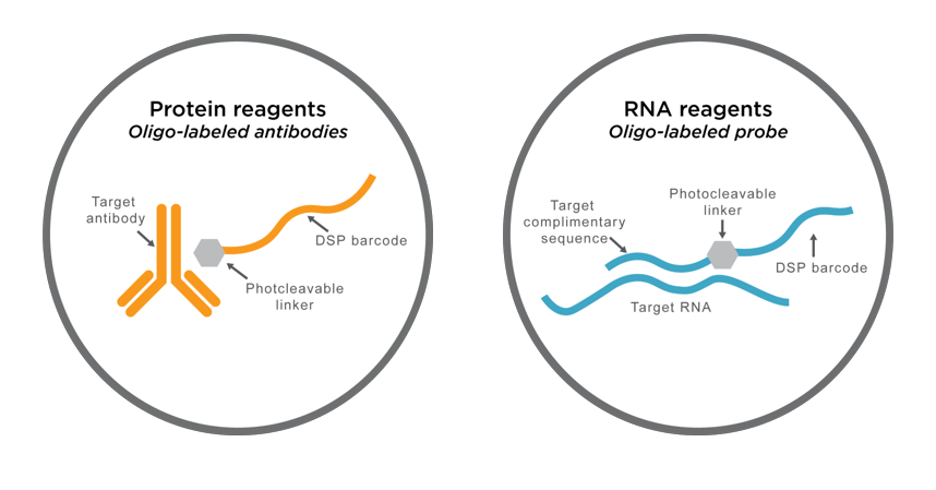
Compañía: NanoString
- Libera barcodes indicadores por exposición UV en la región de interés
- Los barcodes son recolectados y secuenciados
- Permite seleccionar regiones complejas guiadas por inmunofluorescencia
Métodos basados en sm-FISH
La hibridización in situ con sondas radioactivas comenzó a usarse en los años 70s
sm-FISH: Características clave
La resolución espacial sólo limitada por la difracción de la luz (~200 nm)
La sensibilidad y precisión son altísimas: pueden contarse moléculas individuales de RNA por célula
Limitante: ¿Cómo detectar >20,000 genes con sólo <5 colores de fluoróforo?
Múltiples rondas de hibridación?
F = Número de fluoróforos · N = número de rondas
Genes detectables = F × N
seqFISH+: Pseudo-colores
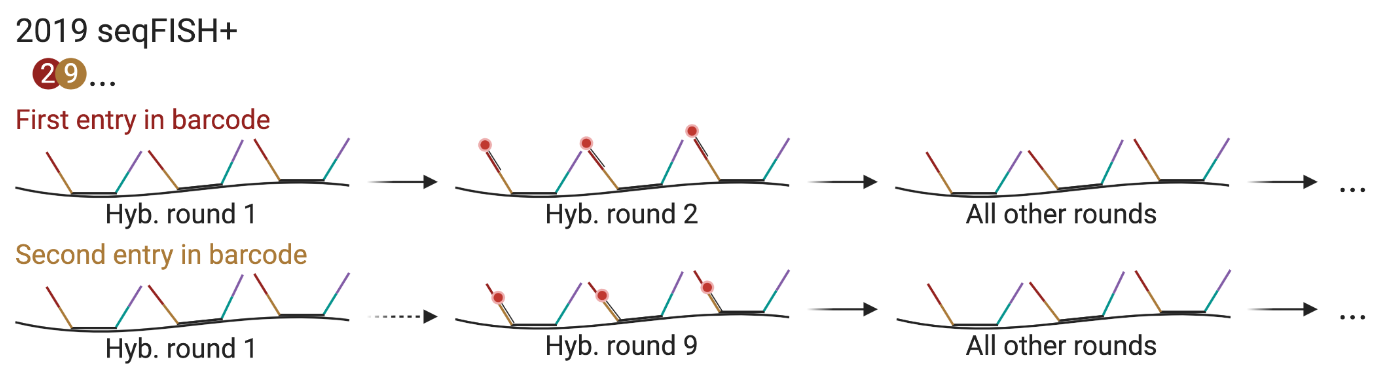
Utiliza “pseudo-colores”: el número de rondas se multiplica con el número de pseudocolores
Por ejemplo, 20 pseudocolores se leen en 20 rondas. Cada gen solo se detecta una vez en 20 rondas. La ronda en la que aparece es su número de barcode
En la segunda “ronda” (21 a 40), se lee el segundo “pseudocolor”.
Posibles genes en 20 rondas con pseudocolores de 3:
\(20^3 = 8000\) o, si se usan 3 diferentes canales/fluoróforos: 24,000Aunque en realidad son 60 rondas! Mejor que sólo 60 o 180 genes, though

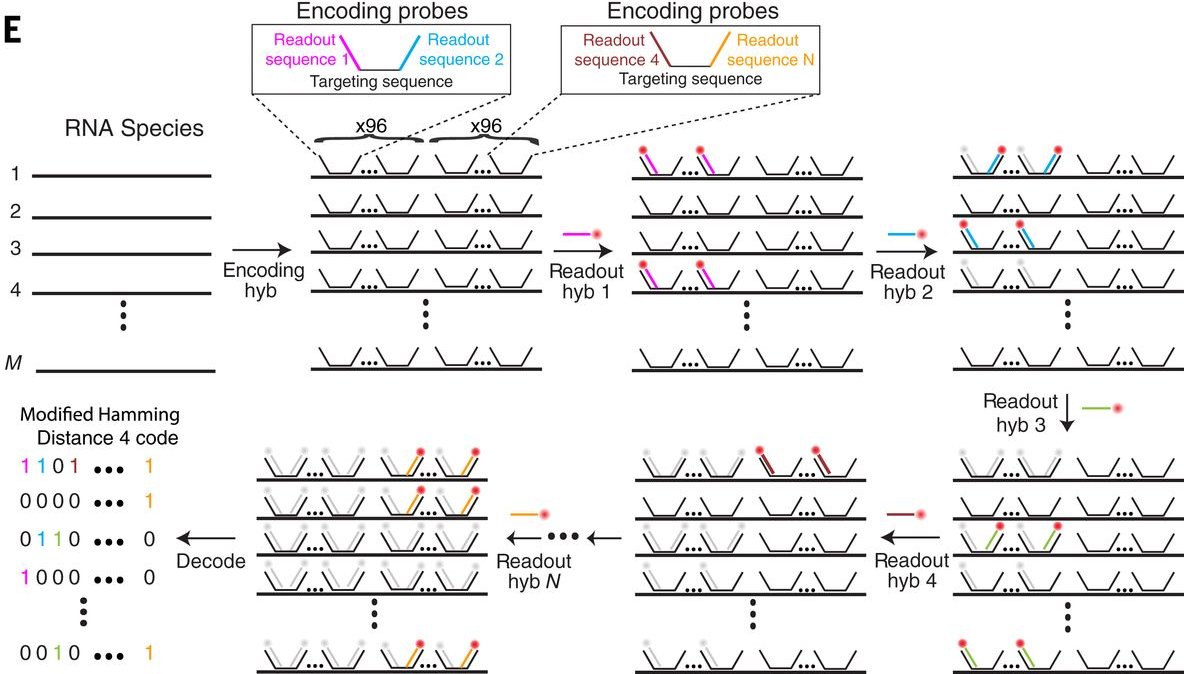
MERFISH: RNAs codificados con palabras binarias
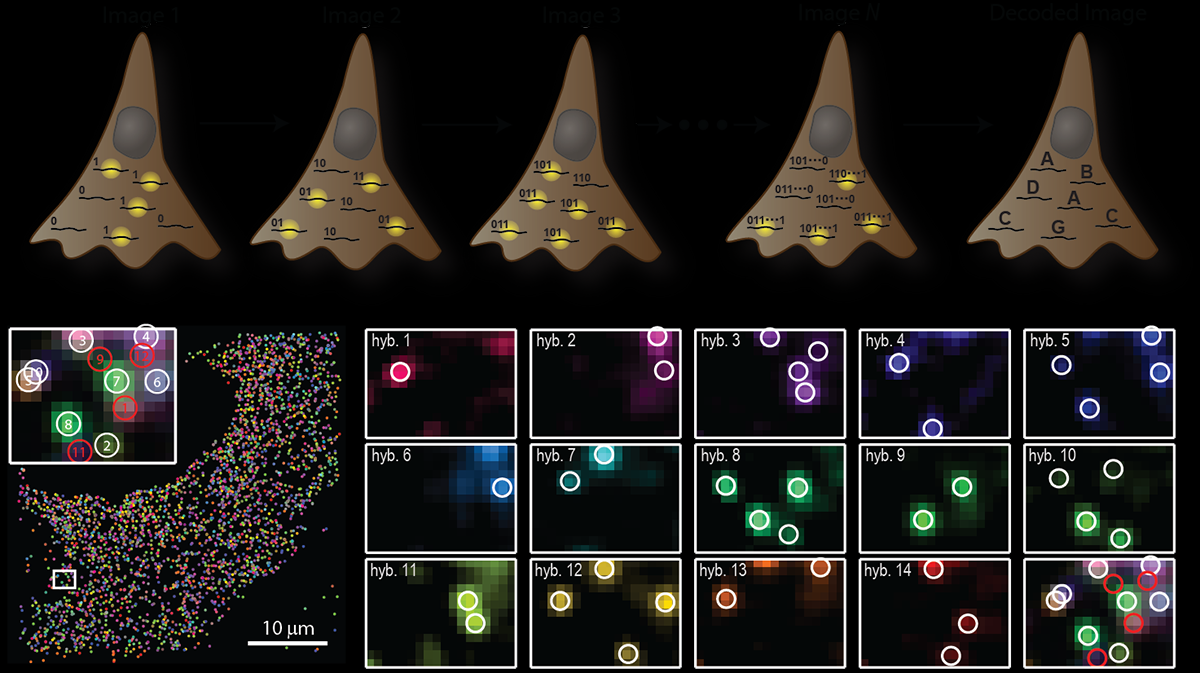
- La ausencia de señal también se toma como información
- RNAs son codificados con palabras binarias a lo largo de múltiples rondas
- Sistema comercial: Vizgen Merscope
MERFISH: Corrección de errores
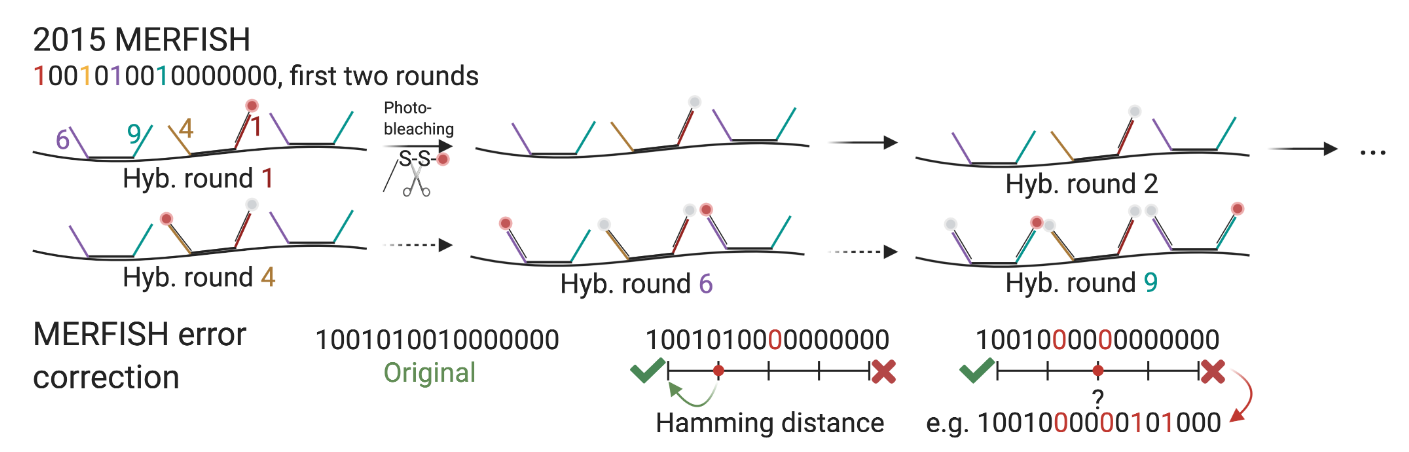
Las palabras binarias están diseñadas con distancia de Hamming suficiente para detectar y corregir errores.
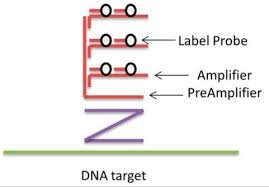
Técnicas de amplificación
Branched DNA
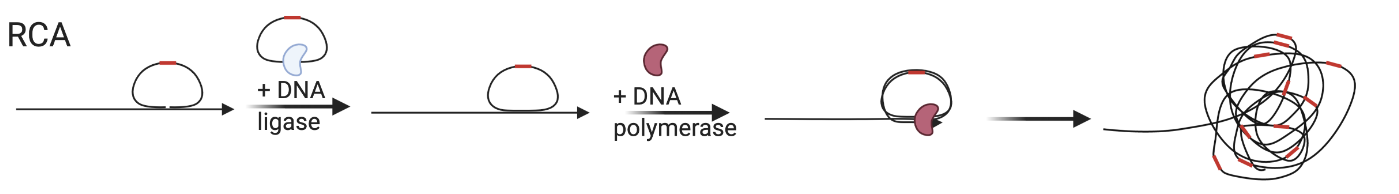
Rolling Circle Amplification
HCR (Hybridization Chain Reaction)
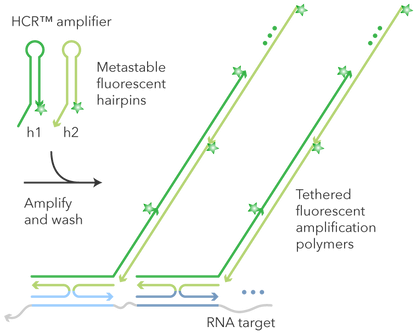
Permiten amplificar la señal fluorescente para mejorar la detección.
Ventajas de métodos smFISH
✅ Altísima sensibilidad: pueden detectar hasta el 100% de transcritos, incluso si son N=1
✅ Resolución de localización sub-celular y en 3D
✅ Se usan como benchmark para comparar la eficiencia de otras técnicas
✅ Disponible información sobre morfología nuclear o celular

Desventajas de métodos smFISH
⚠️ Campo visual pequeño
⚠️ Laborioso y tardado — múltiples rondas de hibridación pueden desprender o dañar el tejido
⚠️ Datos crudos: Terabytes de imágenes en diferentes canales, planos z… Requiere segmentación celular
⚠️ Requieren bombas fluídicas customizadas (ya hay sistemas comerciales)
⚠️ Muchos oligos (oligos con fluoróforos son más costosos); lista pre-definida de genes, típicamente < 300
⚠️ Requieren segmentación celular para asignar transcritos a células y eso es un problema no completamente resuelto…

Secuenciación in situ

Secuenciación por ligación
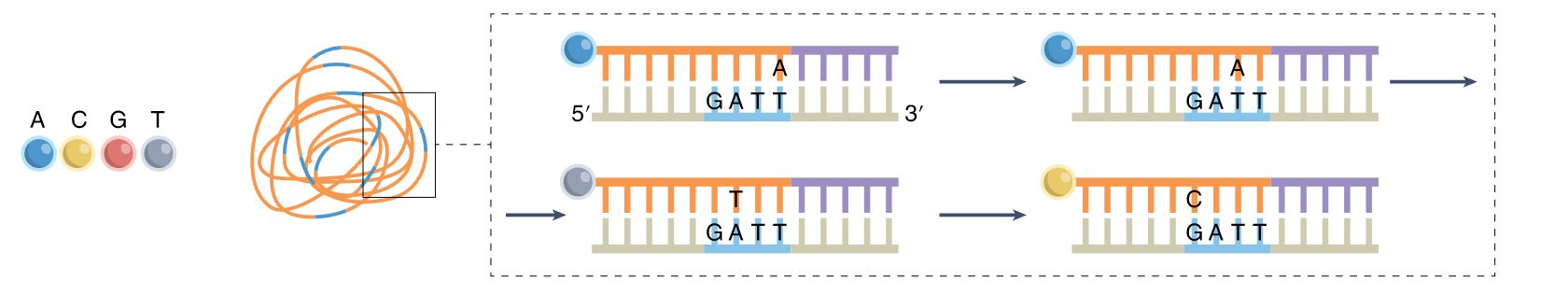
Casi todas necesitan RT pues ligación con RNA es ineficiente.
- SOLiD: el color del fluoróforo codifica las dos bases 3’ con 6 bases degeneradas. Los errores se propagan.
- SEDAL: las regiones que flanquean son conocidas → detección de errores
- cPAL: en la imagen (combinatorial probe anchor ligation)
SOLiD: secuenciación por dinucleótidos
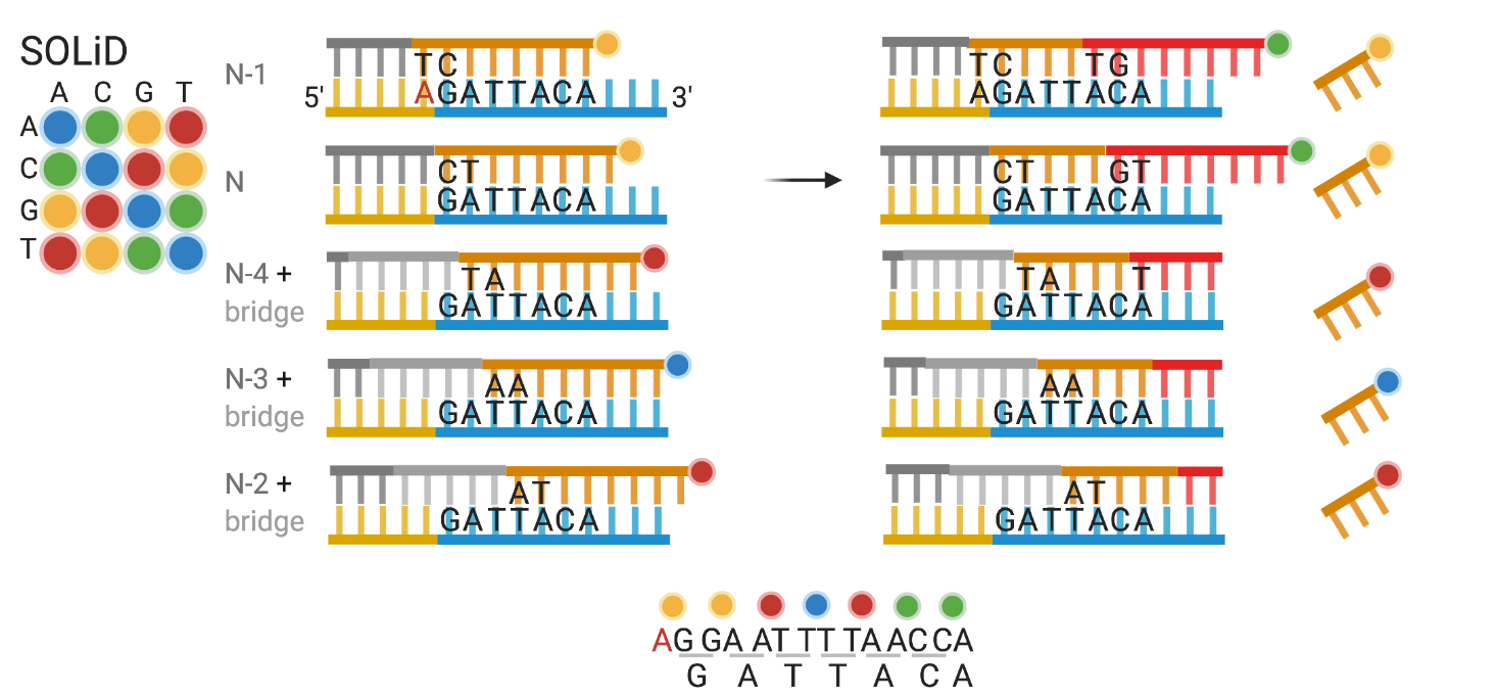
- Moiety que bloquea, sólo una ligación a la vez.
- Cleavage corta 3 nucleótidos junto con el fluoróforo para ligar la nueva sonda
- Para la nueva ronda, se deshibridiza toda la hebra y se comienza con un bridge con un nuevo offset (N-1, N-2, N-3, etc.)
- La A en rojo es parte de una región constante y es conocida.
SOLiD como se usa en FISSEQ
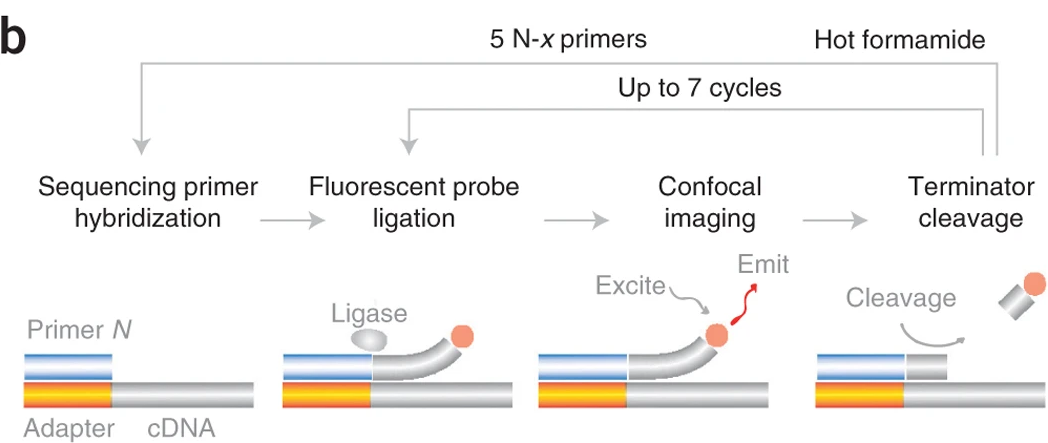
SEDAL & SNAIL probes (Sequencing with error-reduction by dynamic annealing and ligation)
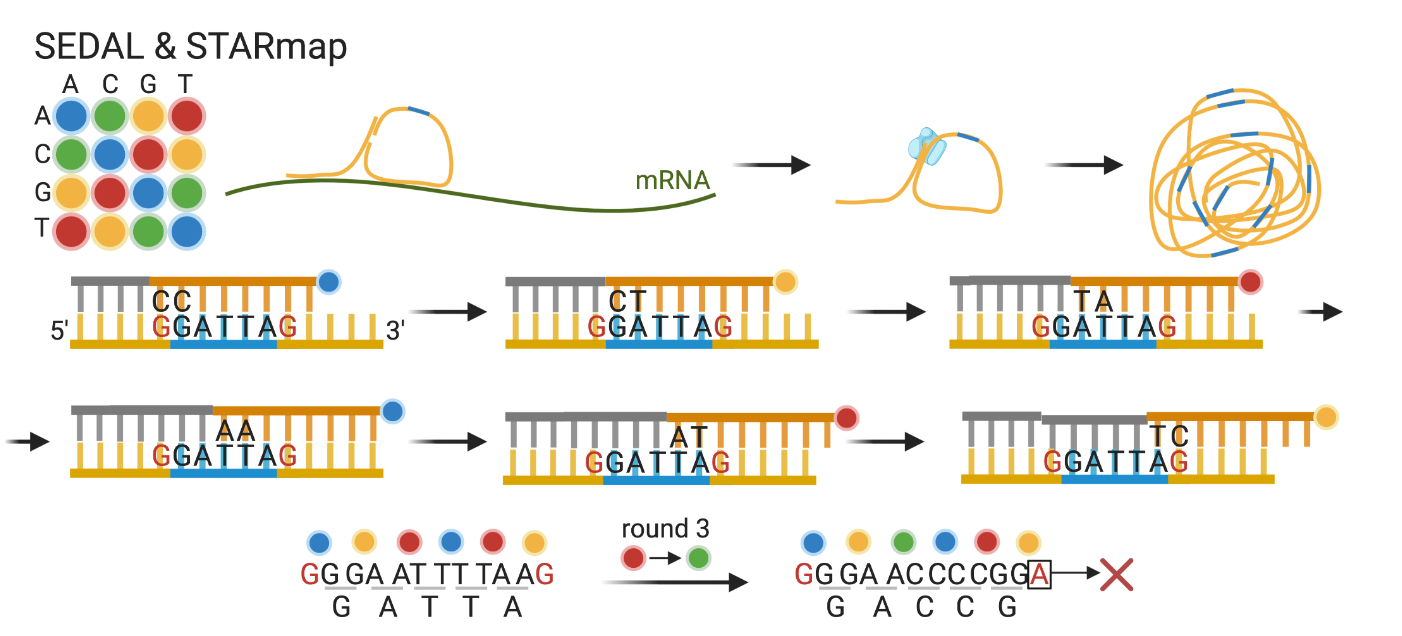
- Specific Amplification of Nucleic Acids via Intramolecular Ligation SNAIL probes: bypass de cDNA synthesis y la ligación ineficiente basada en RNA.
- Sondas con barcode (región azul) especìfico de cada gen.
- Se secuencia el barcode para decodificar el gen blanco.
- Las regiones flanqueantes conocidas permiten detección de errores.
cPAL: combinatorial probe anchor ligation
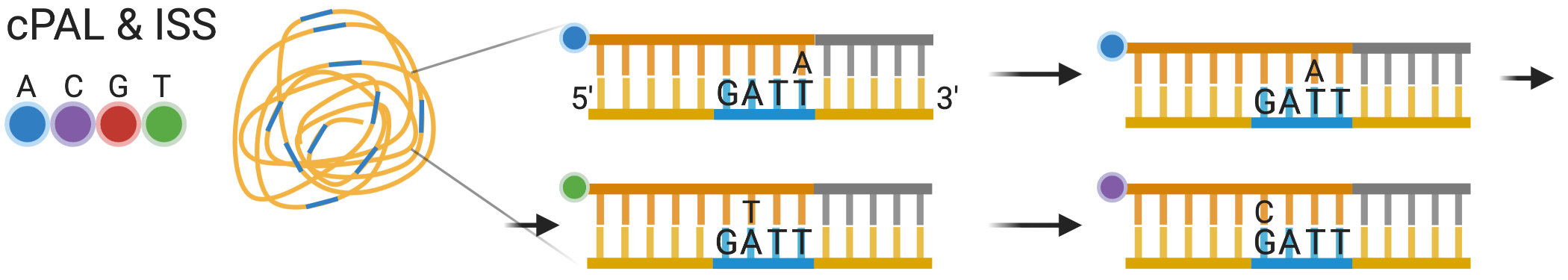
Acá todas las bases son degeneradas excepto la posición que se está interrogando en cada ronda
DBIT-seq: Deterministic Barcoding in Tissue
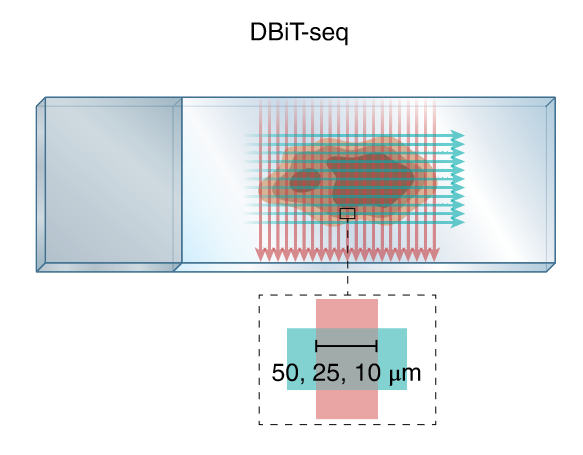
i = 1,2,3…n en coordenada X
j = 1,2,3…m en coordenada Y
- El barcode Aᵢ unido a poly-T se coloca en la fila i (sobre el tejido)
- El barcode Bⱼ unido a UMI, PCR handle, biotina se coloca ortogonalmente
- Barcode Aᵢ y Bⱼ se ligan entre sí y al cDNA
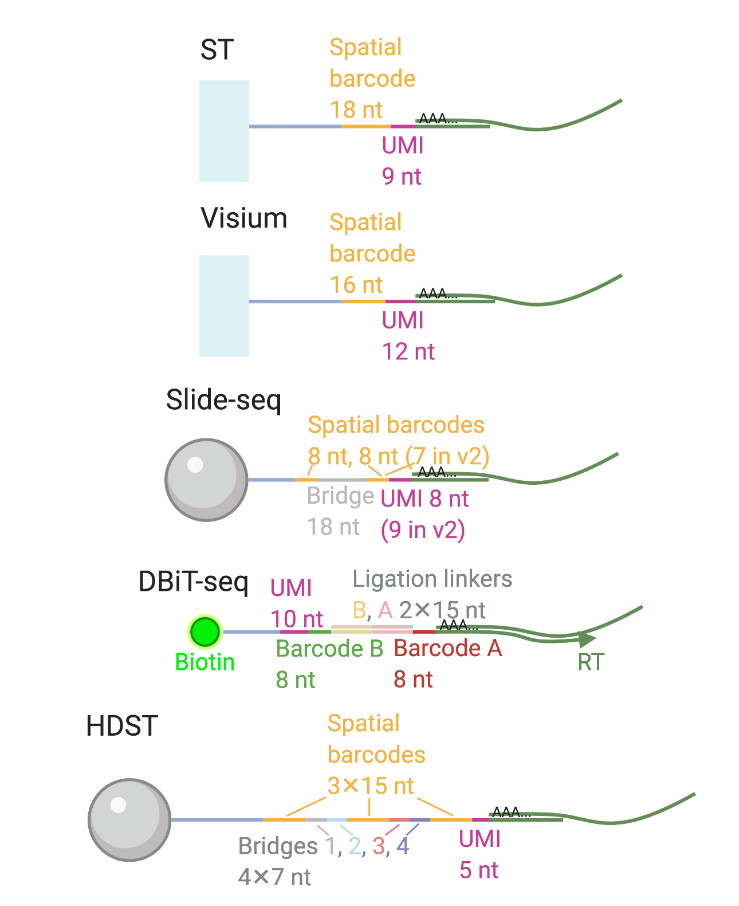
Métodos basados en sm-FISH
- Las perlas se colocan primero y los barcodes se generan por síntesis
- Los barcodes se secuencian con SOLiD o SEDAL
- Slide-seq (Comercializado por Takara como Seeker) y HDST utilizan perlas marcadas
HDST sólo encuentra 1% de los transcritos por área de perla (comparado con smFISH)
Técnicas basadas en arreglos: Resolución en aumento
Los spots de Visium capturan varias células, pero recientemente lanzaron v2.0 con resolución de 2 µm.
La evolución de resolución:
- ST original: ~100 µm
- Visium v1 (A descontinuar en 2026): ~55 µm (varios células por spot)
- Visium HD: ~2 µm (resolución sub-celular)
Nuevas tecnologías a tomar en cuenta:
Illumina está por lanzar su propia tecnología espacial basada en Seq-Scope ? Repurposing Illumina sequencing flow cells for high-resolution spatial transcriptomics
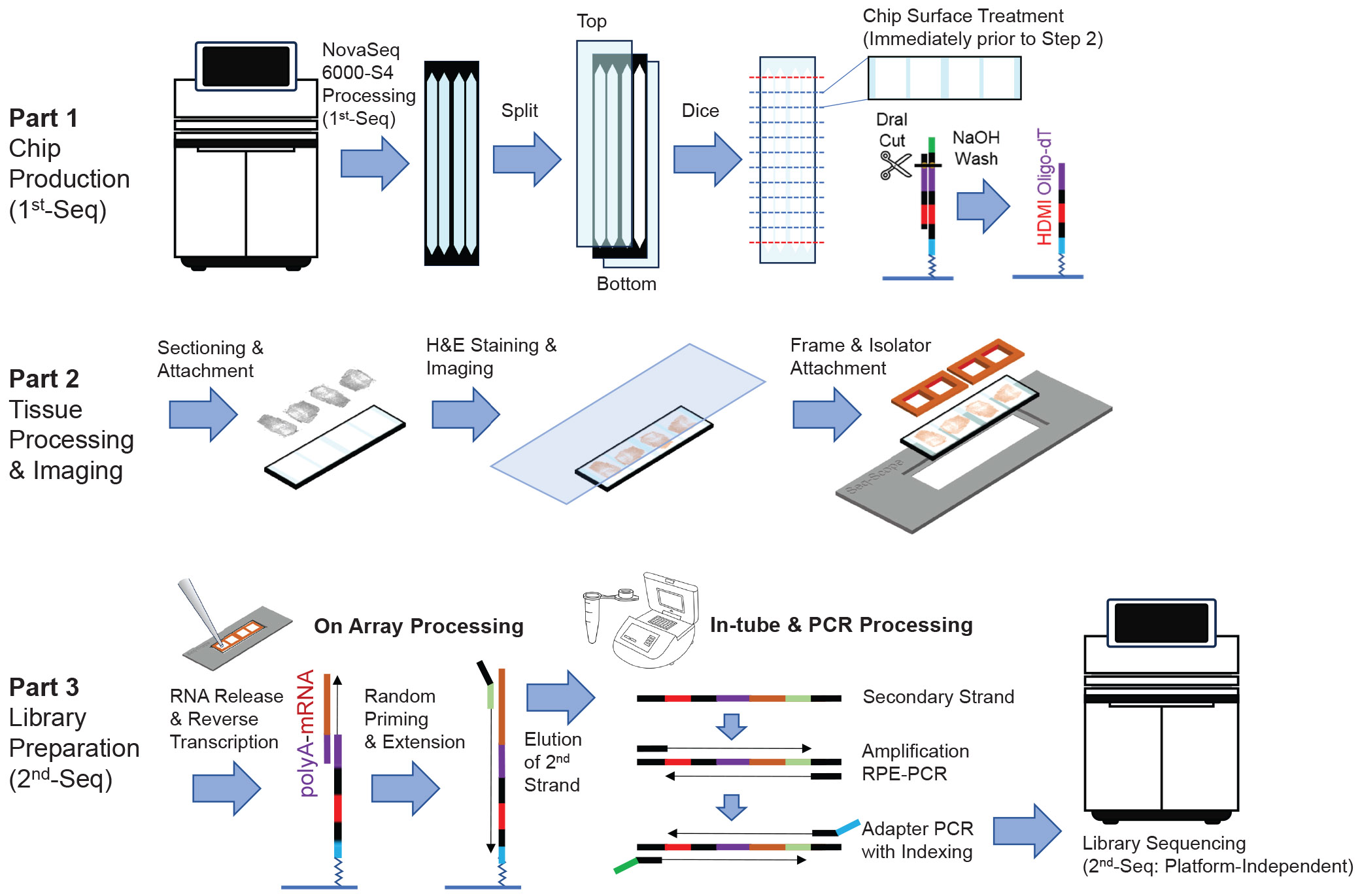
Una idea similar de BGI genomics
Stereo-seq de STOmics
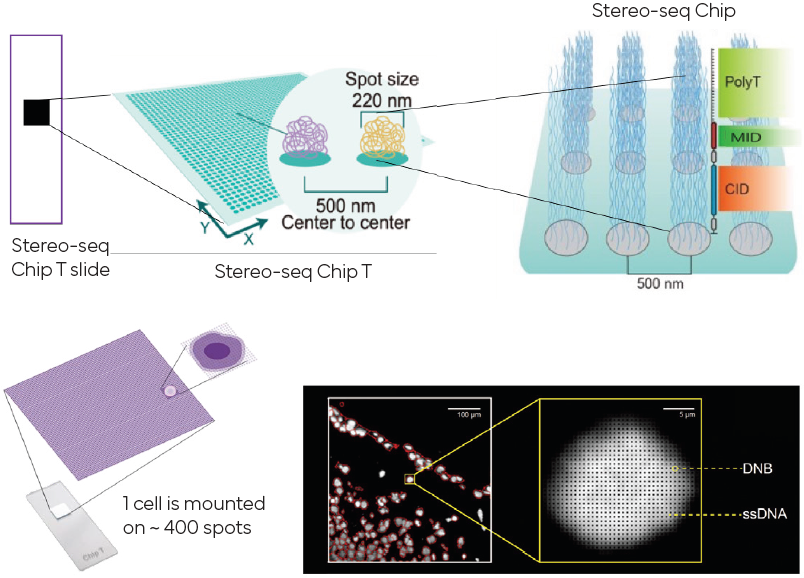
Basado en DNA nanoballs (DNB) dice tener una resolución de 500 nm.
DNA Microscopy: una especie de HiC con transcritos
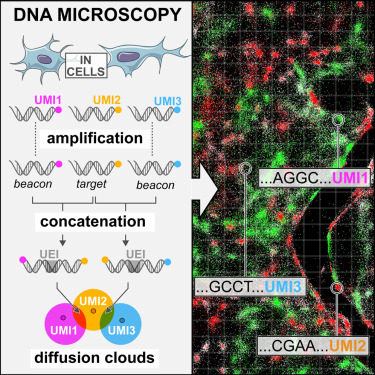
- Transcripción reversa y PCR in situ
- Los productos de PCR (con UMI) se difunden, creando una “nube” alrededor de la molécula original
- Overlap extension PCR concatena cDNAs adyacentes con un UEI (unique event identifier)
- Secuenciación identifica los dos cDNAs, UMIs y evento de sobrelape UEI
- Se reconstruye la distancia relativa entre transcritos: topología vs localización
Trekker: Agrega info espacial a single nuclei
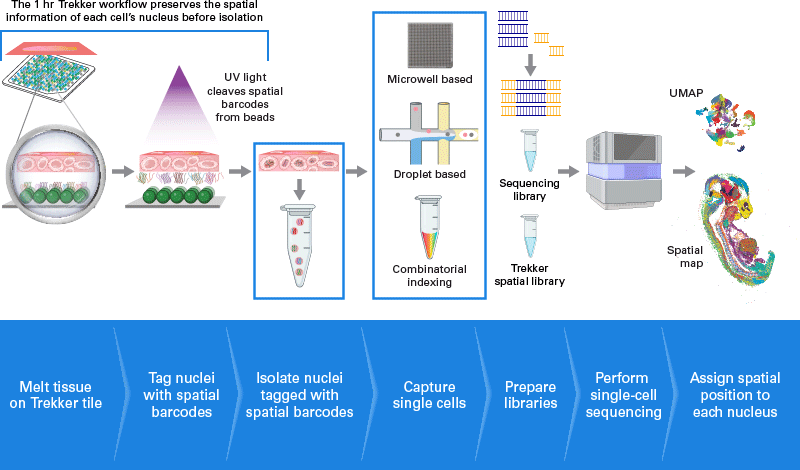
Análisis bioinformático
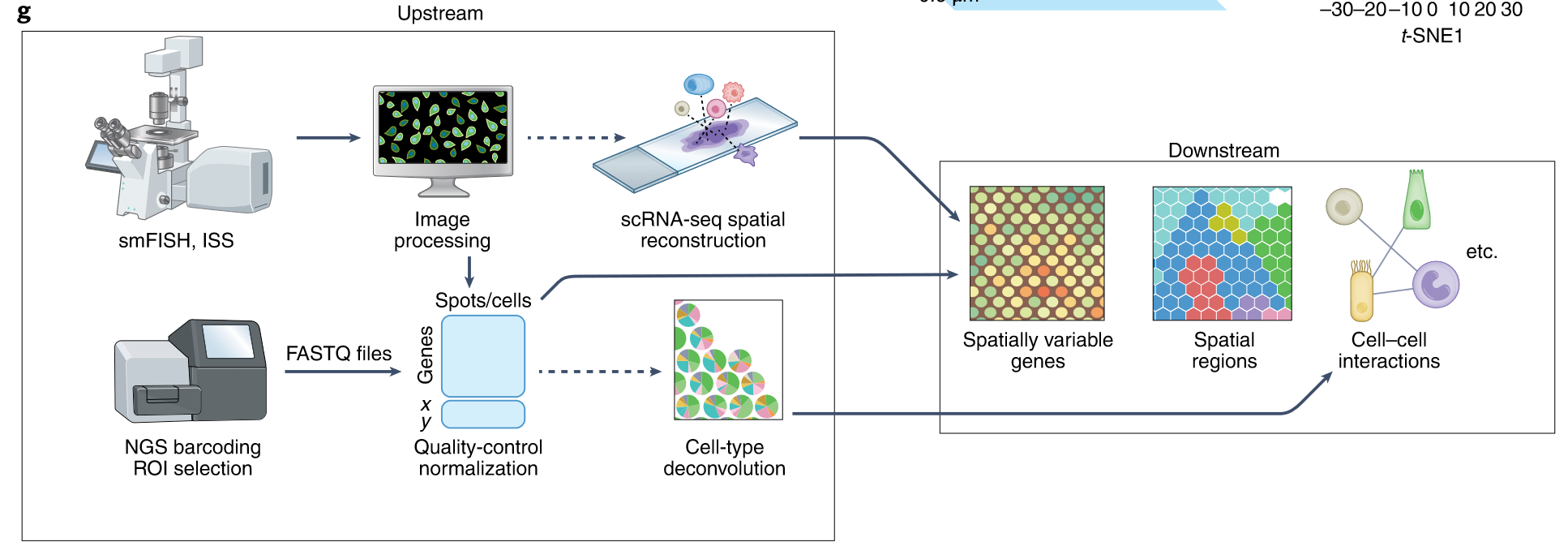
Técnicas smFISH requieren alineación de imágenes de diferentes rondas y segmentación de células a partir de núcleos
Las técnicas estándar de clustering, genes marcadores y expresión diferencial se pueden adaptar a los datos de transcriptómica espacial
La integración de datasets con Seurat se puede usar para hacer deconvolución de scRNA-seq con ST
Paquetes de análisis exploratorio para datos espaciales
Paquetes en R:
- Giotto
- SPATA2
- SEMLA (extiende a partir de objeto Seurat)
Análisis espaciales no disponibles en Seurat:
- Patrones de co-expresión
- Genes espacialmente variables
- Vecindades celulares e interacciones
- Interacciones ligando-receptor

Métodos de reconstrucción espacial de scRNA-seq
- GLISS: Reducción dimensional a 2 o 3D utilizando información espacial (in situs)
- CSOmap: usa información ligando-receptor para reducción de dimensionalidad
- Seurat (2015): integraba scRNAseq con datos de in situ
- LIGER, SpaGE: integración en un espacio latente y proyección de la expresión de scRNAseq a datos espaciales
- En algunos tejidos, el pseudotiempo de diferenciación corresponde al espacio
- Deep learning entrenado con regiones espaciales

Genes variables y patrones espaciales
Encontrar patrones arquetípicos. Clusterización de los patrones génicos.
Re-utilización de una rica tradición de estadística espacial utilizada en estudios geográficos:
- Puntos en el espacio: los puntos están pre-determinados y cada uno tiene un valor
- Datos aéreos: valores agregados para cada ciudad, distrito, etc.
- Raster: rejilla regular sin espacios entre células
- Point-processes: la localización misma de los puntos es la variable a modelar
ST y Visium: combinación de los dos primeros.
Ideas de computer vision, machine learning, estadística multivariada.

Referencias para datos y otros
Spatial Transcriptomics Repository EBI
Museum of Spatial Transcriptomics
